Reconstructing life
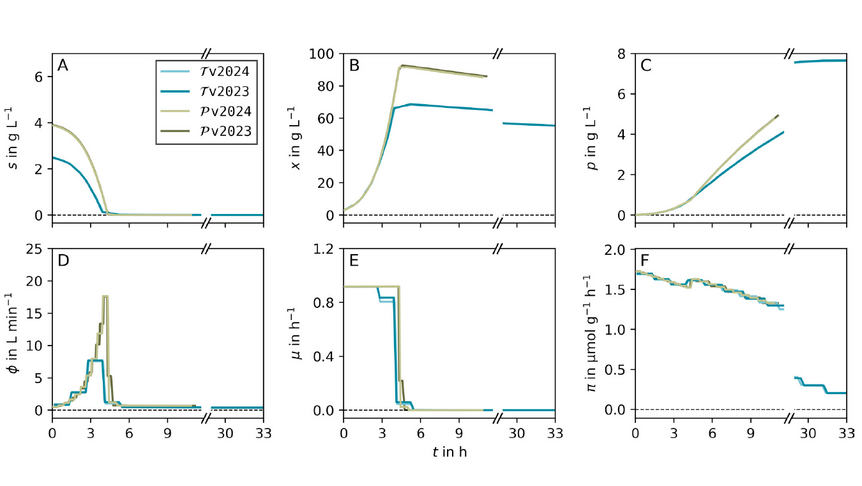
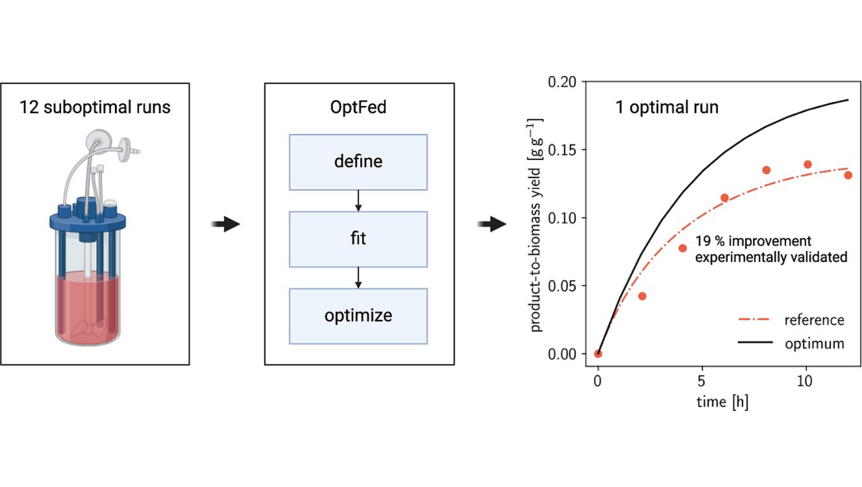
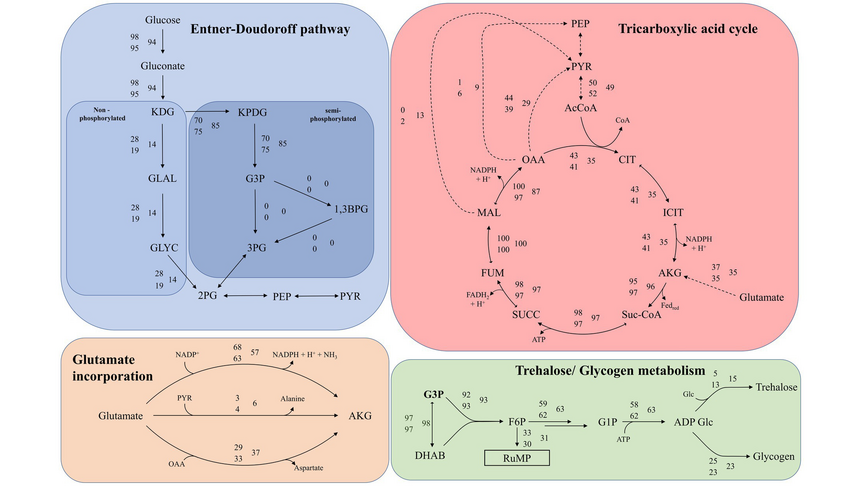
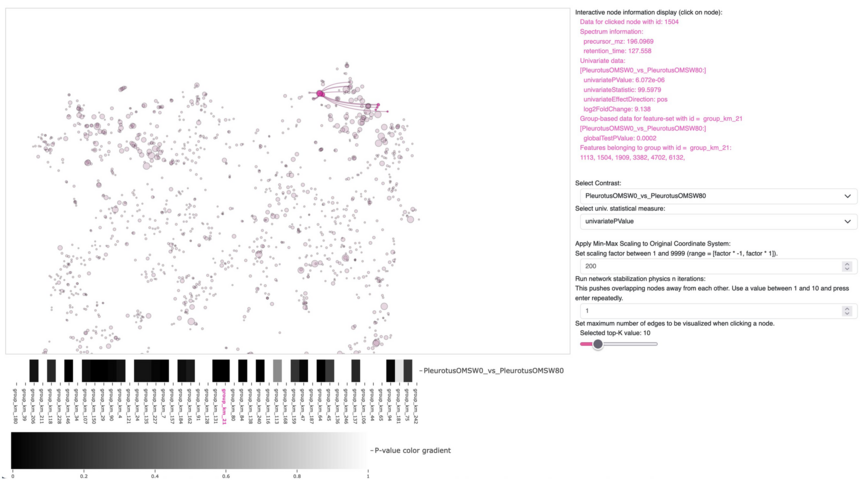
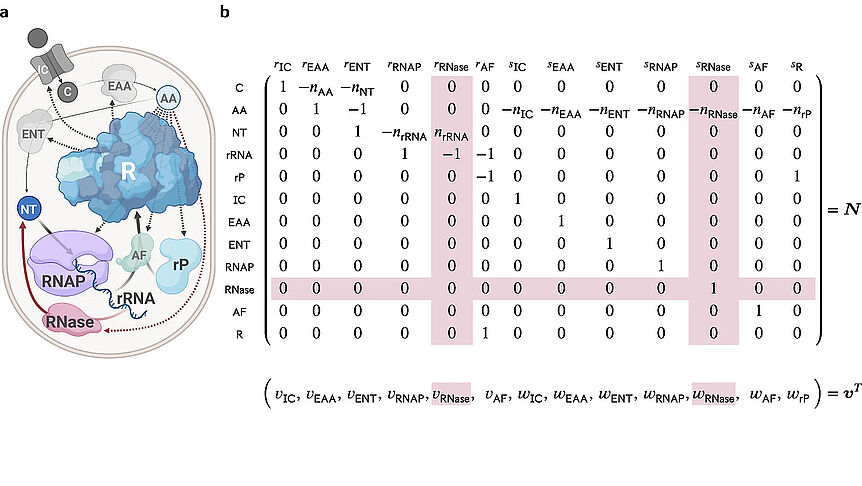
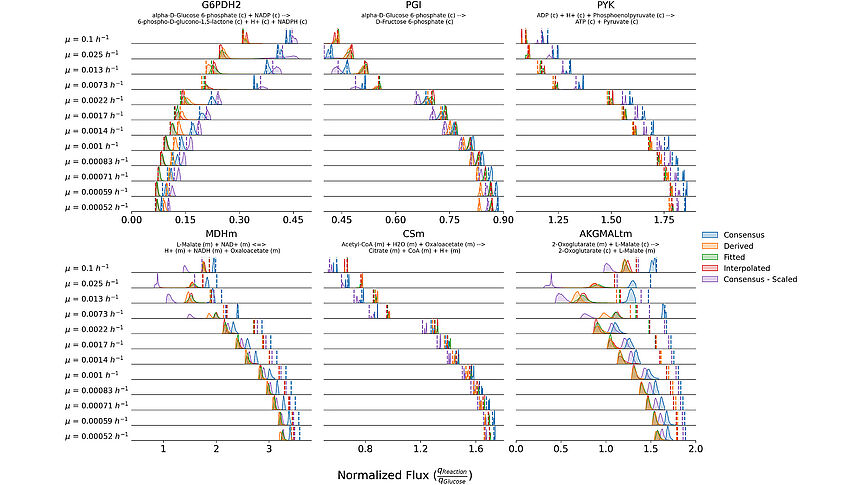
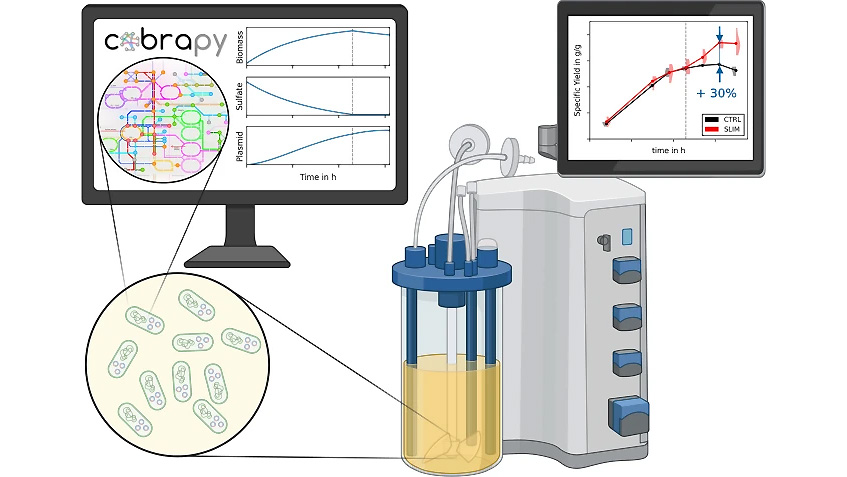
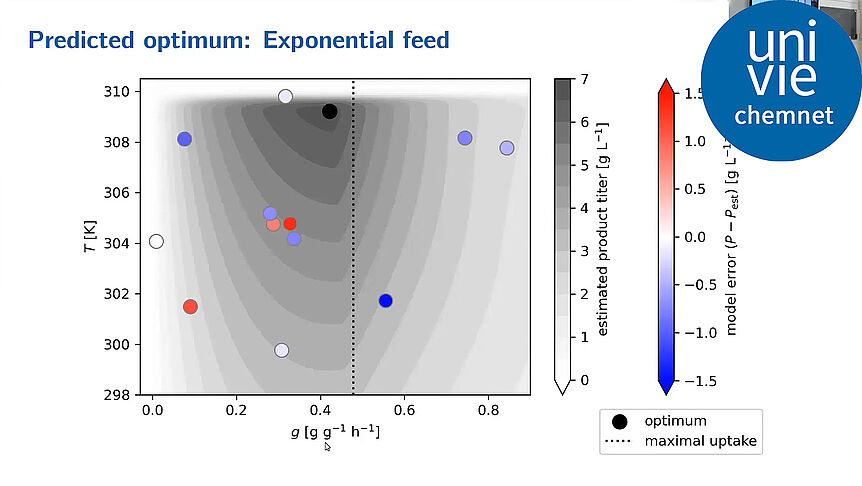
As the knowledge of genes, proteins, and other biological components has developed, so has the interest in studying the interaction between them in order to understand complete biological systems. Here, the systems biology approach provides concepts and tools. The ultimate goal is to rebuild nature on the computer and set up a complete mathematical model of living cells in silico. Such models may then, for instance, be used to optimize the biotechnological production of value-added chemicals by microorganisms.
The research focuses on mathematically modeling the dynamics, regulation, and control of metabolic networks. In particular, we are interested in studying structural, i.e. topological properties of complex metabolic networks and how these structures give rise to metabolic functions. We develop computational tools to predict optimized microbes for biotechnological applications. Most of the experimental work is carried out in collaboration with other research groups.
Cutting-edge research for the future
Cluster of Excellence
Circular Bioengineering
With €16 million in FWF funding, the Cluster of Excellence Circular Bioengineering marks a significant step toward sustainable innovation.
Mission: Cutting-edge research in molecular biosciences and biotechnology to design and engineer bio-based materials that are sustainable, efficient, and waste-free.







 Quantification of SREBP2 n/c ratio in cells exposed to 24 h SS with or without presence of LOVA 1 µM. (B) Quantification of YAP1 n/c ratio in cells exposed to 24 h SS with")
.")
 as a function of the dilution rate (h−1) and biomass (106 cells ml−1).")