Published in PLoS Comput Biol 20(6): e1012236.
https://doi.org/10.1371/journal.pcbi.1012236
Abstract
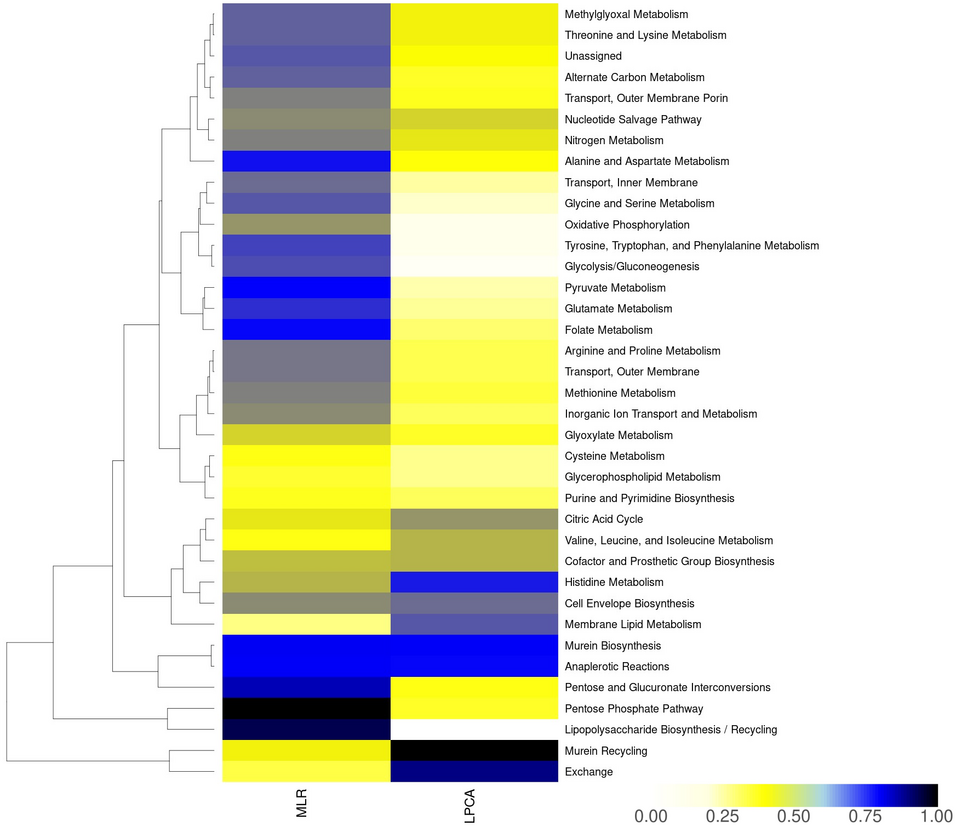
Genome-scale metabolic models (GSMMs) offer a holistic view of biochemical reaction networks, enabling in-depth analyses of metabolism across species and tissues in multiple conditions. However, comparing GSMMs against each other poses challenges as current dimensionality reduction algorithms or clustering methods lack mechanistic interpretability, and often rely on subjective assumptions. Here, we propose a new approach utilizing logisitic principal component analysis (LPCA) that efficiently clusters GSMMs while singling out mechanistic differences in terms of reactions and pathways that drive the categorization. We applied LPCA to multiple diverse datasets, including GSMMs of 222 Escherichia-strains, 343 budding yeasts (Saccharomycotina), 80 human tissues, and 2943 Firmicutes strains. Our findings demonstrate LPCA’s effectiveness in preserving microbial phylogenetic relationships and discerning human tissue-specific metabolic profiles, exhibiting comparable performance to traditional methods like t-distributed stochastic neighborhood embedding (t-SNE) and Jaccard coefficients. Moreover, the subsystems and associated reactions identified by LPCA align with existing knowledge, underscoring its reliability in dissecting GSMMs and uncovering the underlying drivers of separation.
Author’s summary
GSMMs are comprehensive representations of all the biochemical reactions that occur within an organism, enabling insights into cellular processes. Our study introduces LPCA to explore and compare these biochemical networks across different species and tissues only based on the presence or absence of reactions, summarized in a binary matrix. LPCA analyzes these binary matrices of specific biochemical reactions, identifying significant differences and similarities. We applied LPCA to a range of datasets, including bacterial strains, fungi, and human tissues. Our findings demonstrate LPCA’s effectiveness in distinguishing microbial phylogenetic relationships and discerning tissue-specific profiles in humans. LPCA also offers precise information on the biochemical drivers of these differences, contributing to a deeper understanding of metabolic subsystems. This research showcases LPCA as a valuable method for examining the complex interplay of reactions within GSMMs, offering insights that could support further scientific investigation into metabolic processes.